Enlarge Image
Next-Generation Sequencer applications: Innovation to turn ‘junk DNAs’ into genetic markers
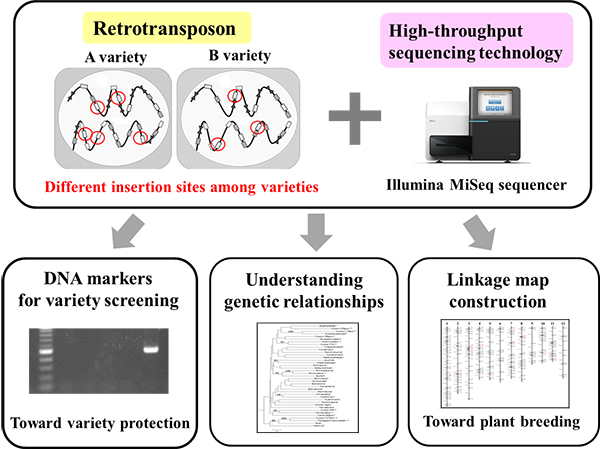
A series of the Next-Generation Sequencer (NGS) applications are being developed to utilize ‘junk DNAs’ as distinctive genetic analysis systems for crop species.
Many nucleic DNAs do not have any known biological functions. Such DNAs are referred to as ‘junk DNAs’. Having accumulated genomic copies throughout evolution, retrotransposons constitute a major portion of junk DNAs. Although most copies have lost copy-making ability, small groups of the retrotransposons still remain competent. The copies of an active group are inserted at markedly different genomic positions, even among closely related varieties of the crop species.
Identification of the active groups is, however, challenging as they are concealed by numerous non-active groups. Additionally, DNA sequences of the numerous positions where active groups are inserted must be efficiently determined for genetic population under study.
Yuki Monden and colleagues at Okayama University, National Institute of Genetics, and Tochigi and Fukuoka Prefectural Agricultural Research Stations have prepared the NGS (HiSeq2000) sequence library of strawberry, which exclusively contains terminal sequences of a major retrotransposon type (LTR: Long Terminal Repeat), and have successfully identified the active groups using novel NGS sequence analysis.
In another study, the insertion site library for two active groups was constructed over 38 sweet potato varieties. The NGS analysis determined 2024 inserted positions, of which 91.4% occurred in a single variety or varieties of different combination.
The series of NGS applications on active retrotransposons provide efficient genetic analysis systems, especially for variety fingerprinting, linkage map construction, and lineage analysis of the crop species [1,2].
Reference1:
・Authors: Yuki Monden, Nobuyuki Fujii, Kentaro Yamaguchi, Kazuho Ikeo, Yoshiko Nakazawa, Takamitsu Waki, Keita Hirashima, Yosuke Uchimura, Makoto Tahara.
・Title of original paper: Efficient screening of long terminal repeat retrotransposons that show high insertion polymorphism via high-throughput sequencing of the primer binding site.
・Journal, volume, pages and year: Genome, (2014).
・Journal website: http://ousar.lib.okayama-u.ac.jp/metadata/52815
・Digital Object Identifier (DOI): 10.1139/gen-2014-0031
・Journal website: http://www.nrcresearchpress.com/doi/abs/10.1139/gen-2014-0031#.U96WD-OKVJl
・Affiliations: Graduate School of Environmental and Life Science, Okayama University.
・Department website: http://www.gels.okayama-u.ac.jp/index_e.html
Reference2:
・Authors: Yuki Monden, Ayaka Yamamoto, Akiko Shindo, and Makoto Tahara.
・Title of original paper: Efficient DNA Fingerprinting Based on the Targeted Sequencing of Active Retrotransposon Insertion Sites Using a Bench-Top High-Throughput Sequencing Platform.
・Journal, volume, pages and year: DNA Research, (2014).
・Journal website: http://ousar.lib.okayama-u.ac.jp/metadata/52816
・Digital Object Identifier (DOI): 10.1093/dnares/dsu015
・Journal website: http://dnaresearch.oxfordjournals.org/content/early/2014/06/13/dnares.dsu015